|
Biochemical/molecular techniques |
Background concepts:
Spectrophotometry and protein concentration estimation |
|
|
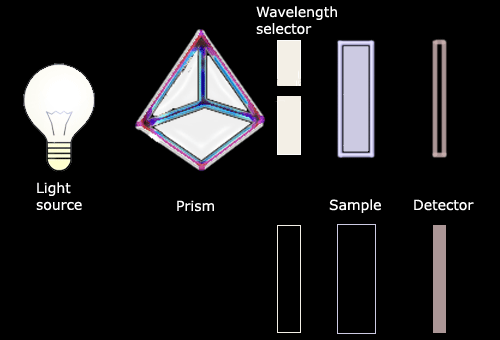
A spectrophotometer works by shining a light beam through a solution
containing an analyte (or molecule being studied). Light absorption is
detected using a photosensitive element which reads out optical density
(O.D.). Estimation of the concentration of the analyte by
spectrophotometry is possible because the quantity of light absorbed by
the analyte in a solution (or O.D.), increases linearly with
concentration. |
|
The technique |
|
The spectrophotometer is a device which
measures the concentration of a substance (analyte) in solution. The
analysis carried out by a spectrophotometer is based on Beer's Law which
relates the amount absorbed by a substance to its concentration in a
solution. The concentration of an analyte is directly proportional to
the amount of light absorbed by the solution, and inversely proportional
to the logarithm of the amount of light transmitted by the solution. |
|
 |
|
 |
|
Beer's law is followed only
if the light entering the solution is composed of a single wavelength.
This light set at a specific wavelength enters the cuvette which
contains the solution to be tested. Some of the incident light (I0)
is absorbed by the solution - the amount absorbed depends on the
concentration of the solution - and the rest of the light (the
transmitted light I) is detected. The ratio between the amount of light
that goes into the solution and the amount of light that leaves the
solution gives the absorbance (A) of the solution: A= -log (I/I0).
This definition supposes that all the incident light is either
transmitted or absorbed, reflection or scattering being negligible.
The following equation: A=elc ,
where e
is the substance and wavelength specific absorption coefficient,
I is the length the light travels through the sample
and c is the concentration of the sample,
shows the relationship between the absorbance and the concentration of a
substance. |
|
The standard curve |
|
Since the samples are tested
under the same condition: under a set wavelength (which is the
analytical wavelength: the wavelength of maximal absorbance chosen from
the absorbance spectrum of the substance) and incident light distance,
e
and
l are the same for all samples, thus A varies linearly with c.
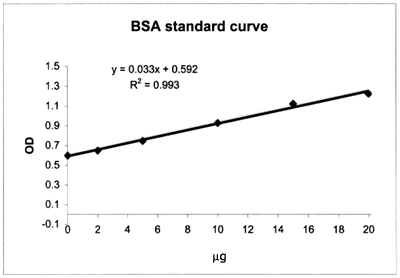
The first step is to construct a curve relating known analyte quantities to their measured absorbance. OD recorded from the
spectrophotometer (absorbance) is plotted on the Y-axis and the
analyte amounts are plotted on the X-axis. A series of solutions with
known analyte quantity (the "standards") are prepared, the
absorbance values are recorded and plotted versus the solution
amounts; a straight line can be best fitted. The first
spectrophotometer cuvette is the "blank": it contains no analyte, the OD
is recorded.
|
|
|
Example of a calibration
curve constructed with a set of Bovine Serum Albumin (BSA) standards;
note that the first standard contains all reagents except the protein (BSA):
|
Absorbance
(OD) |
Quantity
BSA (mg) |
| 0.598 |
0 |
| 0.648 |
2 |
| 0.745 |
5 |
| 0.927 |
10 |
| 1.123 |
15 |
| 1.225 |
20 |
|
|
Next, the absorbance of the unknown samples is measured.
The absorbance value will correspond to a value on the
calibration graph or standard curve. |
|
The Bradford method of
protein quantification |
|
Assays which generate a
protein-dependent colour change, are measured by spectrophotometry:
Beer's law can be applied here for accurate quantitation of protein by
selecting an appropriate ratio of dye volume to sample concentration.
Over a broad range of protein concentrations, the dye binding method
gives an accurate but not entirely linear response.
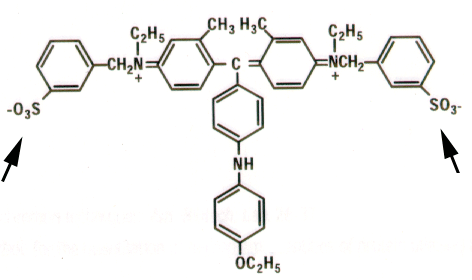
The Bradford assay is
mediated by the Coomassie Blue G-250 dye.
As with any
colorimetric protein assay, there is variation in colour response to
different proteins. Both the colour change due to dye binding and the
variation in colour response can be attributed to the dye having three
absorbing species: a red cationic species, a green neutral
species, and a blue anionic species. At the assay pH (under strongly
acidic conditions) the dye molecules are doubly protonated and are
present as the red cationic dye form. Binding of the dye to protein
stabilizes the blue anionic dye form, detected at 595 nm.
|
 |
|
Coomassie
brilliant Blue G-250: the higher degree of conjugation (single/double
bonds), the higher the absorption of light.
The Coomassie blue reagent has been shown to interact mainly with
arginine residues, but weakly with histidine, lysine, tyrosine,
tryptophan and phenylalanine residues; VanderWaals forces and
hydrophobic interactions also participate in the binding mechanism.
The number of Coomassie reagent ligands bound to each protein molecule
is approximately proportional to the number of positive charges on the
protein (1.5 - 3 dye molecules/charge). |
|
The assay is also influenced
by non-protein sources (e.g.: detergents) and becomes progressively more
non-linear at the high end of its useful protein concentration range:
interference from non-protein compounds is due to their ability to shift
the equilibria among the three species of the dye. The assay is also
protein-dependent (different proteins have different amino acid
compositions) and varies with the composition of the protein.
These limitations result in the necessity of having the appropriate
protein standard solution; to get the most accurate results, the
standard must be composed of a mixture of proteins as similar as
possible to the unknown. Bovine gamma globulin or bovine serum albumin
may be used as standards. The standard will give a colour yield similar to the protein
being assayed. The assay can be used with samples having protein
concentrations between 2-20 mg/ml. |
|
|
|
|
|
|